
1研究背景与目的
发育障碍是一组复杂的疾病,严重影响患者的生活质量,给家庭和社会带来沉重负担。随着高通量测序技术的发展,虽然在诊断方面取得了一定进展,但仍有许多患者病因不明。常染色体隐性编码变异被认为是发育障碍的重要病因之一,但此前研究在样本量和群体多样性上存在局限,影响了对其全面理解。
本研究纳入了来自DDD研究的13,450个家系和GeneDx的36,057个样本,共计29,745个三联体(trios),其中20.4%具有非欧洲遗传血统,这是目前该领域规模较大且具有丰富遗传多样性的样本集。本研究旨在整合大规模、多血统的样本数据,深入探究常染色体隐性编码变异在发育障碍中的贡献,明确已知基因和变异的解释程度,并寻找新的致病基因,为临床诊断和治疗提供更有力的依据。
2研究方法与过程
2.1样本来源与选择
2.1.1 DDD研究样本
2011年4月至2015年4月期间招募,共13,450名患者(88%为三联体),均患有严重发育障碍且经过常规临床遗传学检查未确诊。纳入标准涵盖神经发育障碍、先天性异常、生长异常、畸形特征和异常行为表型等。研究获得了患者家庭的知情同意,并得到相关研究伦理委员会的批准。
2.1.2 GeneDx样本
患者因疑似孟德尔遗传病被转诊进行临床全外显子测序。根据患者医疗记录中与“神经系统异常”相关的716个HPO术语进行筛选,确保样本具有相似的研究基础。研究遵循相关伦理准则,患者在进行基因检测时已获得知情同意,且数据使用获得了授权。
2.2数据分析流程
2.2.1确定遗传血统(GIA)群体
为了探索不同群体间的遗传结构差异,首先对样本进行GIA分类。通过分析1000 Genomes和Human Genome Diversity Panel参考数据集中的常见单核苷酸变异(MAF>0.01且缺失率<10%),计算样本的主成分(PCs)。利用这些PCs将样本投影到参考数据集的PC空间中,通过UMAP和HDBSCAN聚类算法,定义了六个大陆级GIA群体(AFR、AMR、EAS、EUR、MDE、SAS)和47个精细尺度的GIA亚组,从而实现了在不合并个体水平遗传数据的情况下,准确识别样本的遗传血统。
2.2.2数据质量控制与变异过滤
对两个队列的外显子数据进行联合质量控制,包括变异水平、基因型水平和样本水平的质量控制。例如,在变异水平上,保留MAF≤0.005(在所有GIA亚组中且在gnomAD v2.1.1 GIA群体中)且无纯合基因型(在gnomAD中)的变异;去除与已知近期片段重复或简单串联重复重叠以及不在两个队列外显子捕获试剂盒交集区域内的变异。同时,对变异进行注释,将其分为同义、LoF和功能三类,并进一步对功能变异进行筛选,以确保分析的准确性。
2.2.3负担分析
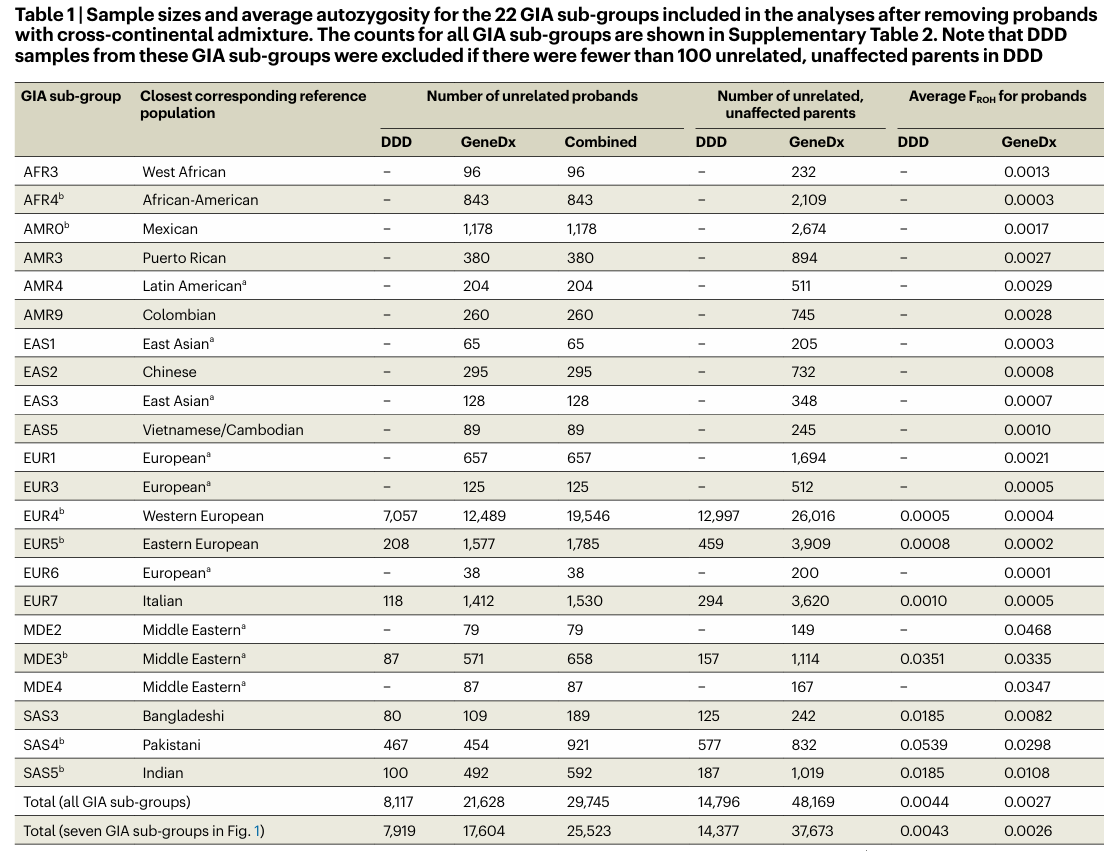
计算不同队列和GIA亚组中罕见双等位基因型(homozygous non - reference或compound heterozygous genotypes)的预期概率,考虑了四种基因型后果类别(同义/同义、LoF/LoF、LoF/功能性、功能性/功能性)。通过比较观察到的和预期的双等位基因型数量,确定每个GIA亚组中可归因于常染色体隐性编码变异的患者比例(即“归因分数”)。为确保分析的可靠性,重点关注了七个大型GIA亚组(AFR4、AMR0、EUR4、EUR5、MDE3、SAS4和SAS5),这些亚组具有足够的样本量且在严格的质量控制下表现稳定。
2.2.4基因发现
对每个基因进行有害双等位基因型的富集测试,以识别新的ARDD基因。采用Poisson检验比较每个基因在不同GIA亚组中观察到的和预期的双等位基因型总数,考虑了四种可能的有害基因型组合,并取每个基因的最低P值进行分析。使用Bonferroni校正(P < 7.2×10⁻⁷)和错误发现率(FDR)控制(FDR < 5%)来确定显著基因,同时通过计算患者间HPO术语的语义相似性分数来评估基因与表型的相关性。
2.3关键数据支撑
在确定GIA群体时,使用了17,693个单核苷酸变异进行分析,计算了样本的前20个主成分,通过UMAP可视化展示了六个大陆级GIA群体的分布。
在负担分析中,对于双等位基因型的计算,考虑了基因特异性的等位基因频率、GIA亚组特异性的等位基因频率和平均纯合度水平等因素,确保了计算的准确性。例如,在计算预期频率时,使用了特定的公式(如({\lambda }{c,p,g}=(1-{a}{p,g}){f}^{;2}{!c,p,g}+{a}{p,g};{f}{c,p,g})),其中({f}{c,p,g})是通过计算无关、未受影响父母中含有至少一个特定类型变异(MAF < 0.005)的单倍型频率得出。
3研究结果与分析
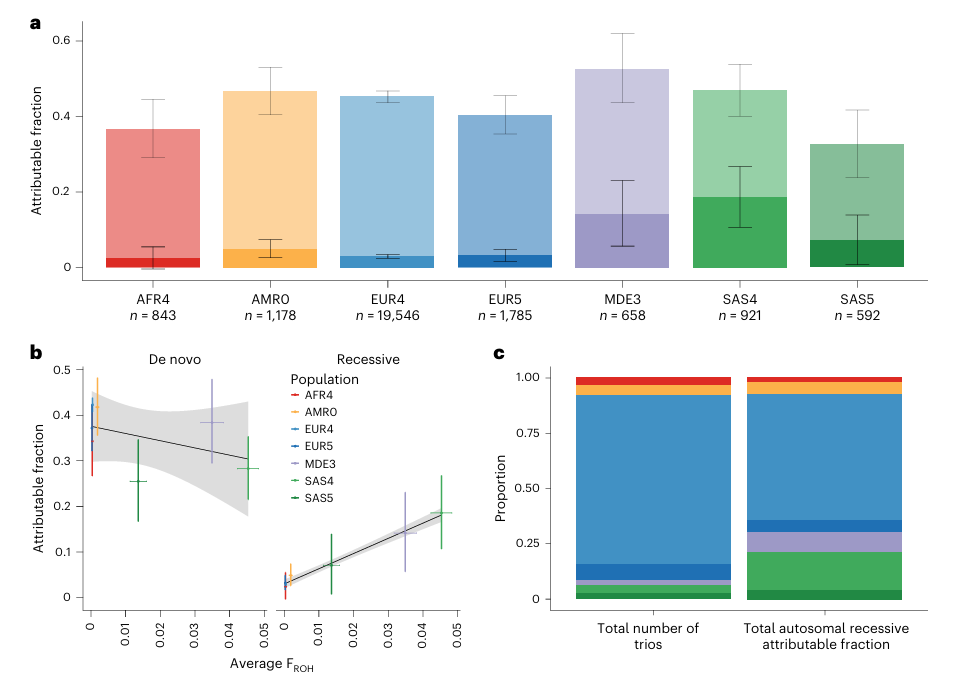
3.1常染色体隐性编码变异的贡献因血统而异
不同GIA亚组中,归因于常染色体隐性编码变异的患者比例差异明显。例如,在SAS4群体中,这一比例高达18.6%(95% CI, 10.7 - 26.7%),MDE3群体为14.1%(5.7 - 23.1%),而在AMR0、EUR4、EUR5和SAS5群体中则相对较低,约为2 - 7%。
常染色体隐性归因分数与平均纯合度水平呈显著正相关(r = 0.99,P = 5×10⁻⁶,七个GIA亚组数据)。这表明在高纯合度群体中,常染色体隐性编码变异对发育障碍的贡献更为突出,如SAS4和MDE3群体虽仅占总样本数的6%,但却占总常染色体隐性归因分数的26.0%。
3.2已知ARDD基因解释了大部分隐性负担
研究中考虑的1,818个已知ARDD基因解释了总常染色体隐性编码负担的84.0%。其中,“共识”基因(在DDG2P和GeneDx列表中均存在的基因)解释了68.1%(62.0 - 74.4%)的负担,“共识 + 不一致”基因(仅在一个列表中的基因)解释了84.0%(76.9 - 91.3%)。
在欧洲血统(EUR4 + EUR5)个体中,已知ARDD基因解释的比例高达86.9%(78.1 - 96.1%),而在非欧洲血统(AFR4 + AMR0 + MDE3 + SAS4 + SAS5)个体中为79.8%(67.9 - 92.3%),两者存在显著差异(P = 0.003),这反映了不同血统群体中已知基因的贡献程度有所不同。
3.3大量潜在诊断可能被遗漏,尤其是错义变异
在未确诊患者中,仍有相当比例可归因于已知ARDD基因中的有害双等位基因编码变异。在DDD未确诊患者中,约1.2%(0.7 - 1.8%)可归因于常染色体隐性DDG2P基因中的此类变异,且均为LoF/功能性或功能性/功能性基因型;在GeneDx未确诊患者中,这一比例为1.6%(1.2 - 1.9%),其中87.8%为LoF/功能性或功能性/功能性基因型。
已知ARDD基因中,约34.4%(27.9 - 41.4%)的隐性归因分数由未在ClinVar中注释为致病或可能致病(P/LP)的变异解释。在DDD中,这一比例更高,达46.7%(32.6 - 62.1%),而GeneDx中为30.3%(23.1 - 38.1%),这凸显了准确解读已知基因中未被充分注释变异的重要性。
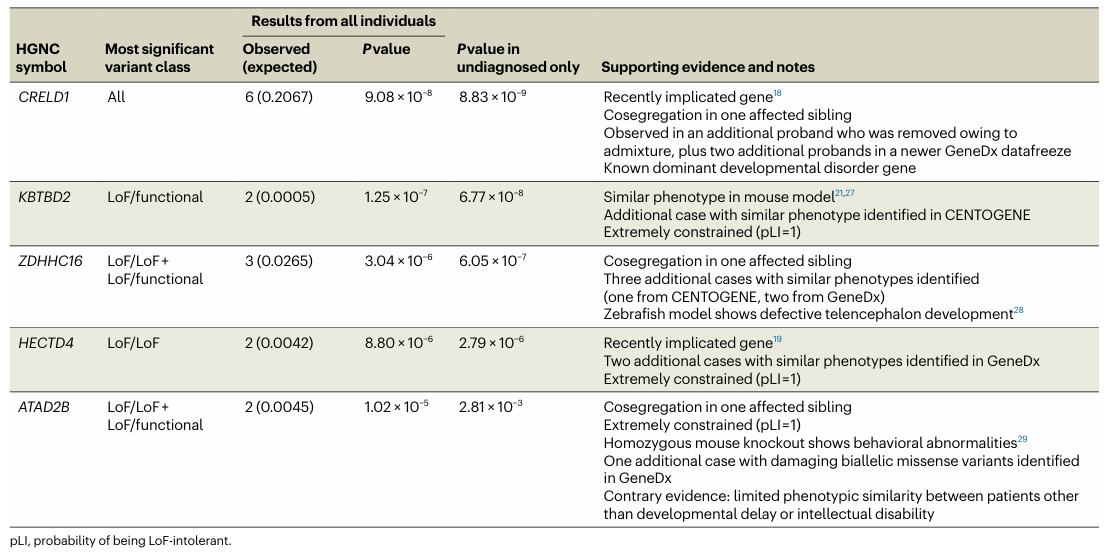
3.4发现两个新的ARDD基因
通过严格的基因负担测试,鉴定出两个新的ARDD基因:KBTBD2和ZDHHC16。在主要基因发现分析中,共有24个基因通过Bonferroni校正,42个基因通过FDR < 5%,其中KBTBD2和CRELD1通过Bonferroni校正,ZDHHC16在仅使用未确诊患者数据进行重复分析时也通过了Bonferroni校正(P = 6.05×10⁻⁷)。
对于KBTBD2基因,观察到两名患者具有破坏性的LoF/功能复合杂合基因型(P = 1.3×10⁻⁷)。该基因编码的蛋白参与胰岛素信号调控,小鼠模型中敲低该基因会导致胰岛素抵抗等表型,与两名患者表现出的生长迟缓等特征部分一致,后续在CENTOGENE数据库中还发现了具有该基因纯合LoF变异且具有相似表型的患者,进一步支持了其致病性。
3.5关键数据支撑
在常染色体隐性编码变异贡献分析中,通过计算不同GIA亚组中各种基因型(如双等位同义基因型)的观察值和预期值,发现大型GIA亚组中两者一致性较好,而小型GIA亚组中观察值常低于预期值,这为样本选择和分析提供了依据(如在七个大型GIA亚组中,总样本量达25,523个无关先证者)。
在基因发现分析中,对每个基因进行了四种有害基因型组合的测试,最终确定了通过严格统计检验的基因,如KBTBD2基因在特定基因型组合(LoF/功能)下的P值极低(1.3×10⁻⁷),表明其在基因水平上的显著异常。
4研究结论与意义
4.1临床诊断方面
已知ARDD基因在诊断中具有重要意义,但目前存在大量未被准确解读的变异,尤其是错义变异。临床医生在诊断过程中,应充分重视已知基因中的潜在致病变异,同时需要更精准的方法来区分致病性和良性功能变异,以提高诊断准确性。
对于未确诊患者,不能忽视已知ARDD基因中可能存在的有害变异,应进一步深入分析这些变异的致病性,避免漏诊。不同基因列表在诊断中的解释能力差异提示应整合多源基因信息,综合判断。
复合诊断中新生突变的潜在贡献不容忽视,对于复杂病例或不完全符合单一基因诊断标准的患者,应考虑进行多基因检测,以全面评估可能的致病因素。
4.2临床治疗方面
新发现的致病基因(如KBTBD2和ZDHHC16)为个性化治疗提供了潜在靶点。深入研究这些基因的功能和致病机制,有望开发出针对性的治疗策略,如针对KBTBD2基因相关疾病,可探索调节胰岛素信号通路的治疗方法。
明确已知基因中未被注释变异的致病性,有助于重新评估现有患者的诊断,进而调整治疗方案。同时,对于具有共同致病机制的ARDD疾病,开发通用治疗策略可提高治疗效率,使更多患者受益。
基于准确的基因诊断,可对患者家族成员进行有效的遗传咨询和筛查,评估生育风险,采取相应的预防措施,如产前诊断或胚胎植入前遗传学诊断,降低疾病在家族中的传播风险。
4.3研究的创新性与局限性
4.3.1创新性
本研究采用了大规模多血统联合分析的方法,整合了来自不同队列的丰富样本,克服了数据治理限制,充分利用了各队列的优势,提高了研究的统计功效,尤其在分析小群体和历史上研究不足的群体方面具有重要意义。
通过严格的统计分析,成功发现了两个新的ARDD基因,并对其进行了初步的功能和表型关联分析,为发育障碍的遗传病因学研究提供了新的线索。
研究不仅关注基因发现,还深入探讨了研究结果对临床诊断和治疗的启示,为推动精准医疗在发育障碍领域的应用提供了重要的理论依据。
4.3.2局限性
研究样本并非发育障碍患者的随机样本,可能存在选择偏倚,导致对常染色体隐性变异的贡献估计不准确,对复合诊断率的评估也可能存在偏差。
在估计归因分数时,假设每个超过预期的有害双等位基因型完全解释一个患者(即完全外显的单基因病因),这可能过于简化了遗传结构,实际情况可能更为复杂。
尽管样本具有一定的祖先多样性,但仍不能完全代表全球人群,且许多GIA亚组样本量较小,影响了对归因分数的精确估计和基因发现的能力。
研究仅关注了蛋白质编码单核苷酸变异和小插入缺失,未包括非编码变异或拷贝数变异的隐性贡献,可能低估了常染色体隐性变异对发育障碍的总体影响。
4.4关键数据支撑
在评估研究局限性时,提到样本选择可能存在偏倚,如在分析未确诊患者中隐性变异贡献时,由于样本选择问题可能导致对潜在诊断遗漏情况的估计不准确。例如,DDD研究中可能遗漏了一些具有隐性条件且容易诊断的家庭,从而影响了对整体隐性变异贡献的评估。
在讨论归因分数估计的局限性时,指出由于假设的简化,实际情况中可能存在其他遗传因素(如多基因或寡基因因素)影响疾病发生,这与研究中观察到的一些现象(如部分患者单一诊断不明确但存在潜在复合诊断因素)相呼应。
5未来研究方向展望
进一步扩大样本量,尤其是针对遗传孤立社区或近亲结婚家庭等特殊群体进行更有针对性的采样,以发现更多的致病基因和变异,更全面地解析发育障碍的遗传结构。
开发更先进的技术和方法来准确解读变异的致病性,特别是错义变异等功能变异。例如,利用深度学习算法或大规模功能实验来评估变异对蛋白质功能和生物过程的影响,提高诊断的准确性。
深入研究复合诊断的机制,明确多种遗传因素(包括新生突变、隐性变异等)在疾病发生发展中的复杂相互作用,建立更精准的复合诊断模型,为临床治疗提供更有效的指导。
开展多中心、跨国合作研究,整合更多不同种族和地区的样本,构建更具代表性的全球发育障碍遗传图谱,以更好地理解不同人群中疾病的遗传异质性和共性。
加强基因 - 环境相互作用的研究,探讨环境因素如何影响常染色体隐性编码变异的表达和疾病的发生发展,为疾病预防和干预提供新的思路。
推动研究成果向临床应用的转化,开发基于新发现基因和变异的诊断试剂盒、治疗药物或干预措施,实现从基础研究到临床实践的有效衔接,改善患者的预后。
参考文献:Nat Genet. 2024 Sep 23. doi: 10.1038/s41588-024-01910-8